library( tidyverse)First, I took the new data and made in index and then verged it with the previous VCF files (from here). I set up filtering to have a minimum QUAL score of 20, no insertion/deletions, minimum allele count of 5 indiviudals posessing the alternative allele state to have the loci be considered, and only loci with 2 alleles present.
#| eval: false
$ bgzip TotalRawSNPs.vcf
$ tabix -p vcf TotalRawSNPs.vcf.gz
$ vcf-merge JK.vcf.gz P.vcf.gz R.vcf.gz S.vcf.gz W.vcf.gz TotalRawSNPs.vcf.gz > FirstRun.vcf
$ vcftools --vcf FirstRun.vcf --remove-indels --minQ 20 --mac 5 --min-alleles 2 --max-alleles 2 --site-depth --out site_depth_summary
VCFtools - 0.1.16
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--vcf FirstRun.vcf
--mac 2
--max-alleles 2
--min-alleles 2
--minQ 20
--out site_depth_summary
--site-depth
--remove-indels
After filtering, kept 1410 out of 1410 Individuals
Outputting Depth for Each Site
After filtering, kept 1055 out of a possible 26395 Sites
Run Time = 6.00 secondsSo, I then looked at the distribution of site depths and found the value to cut off the upper 25% percentile. I then filtered all the sites with depth greater than 20X and 3662X. From that I took a look at the distribution of allele frequencies.
vcftools --vcf FirstRun.vcf --remove-indels --minQ 20 --min-meanDP 20 --max-meanDP 3700 --mac 5 --min-alleles 2 --max-alleles 2 --freq --out first_runThis gives me a distribution of loci whose allele frequencies are:
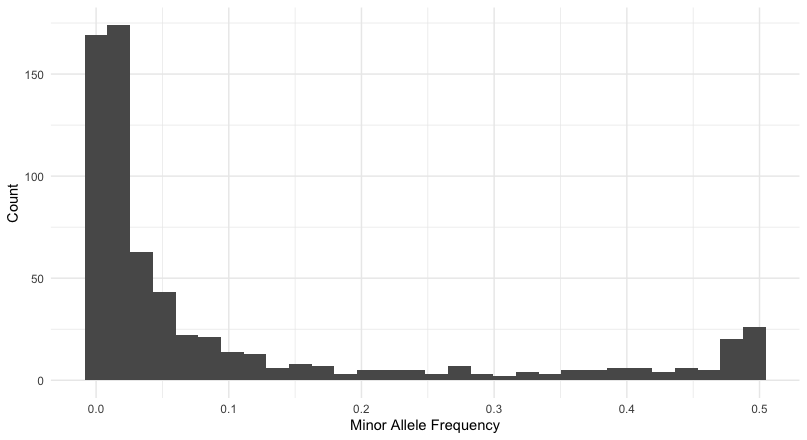
which leaves me with a reasonable number of loci to work with. At present, there are \(663\) loci in the data set and if I were to filter at \(p>=0.05\) that would still leave me with 234 to work with. Now let’s convert these into genotypes and start looking at missing data.