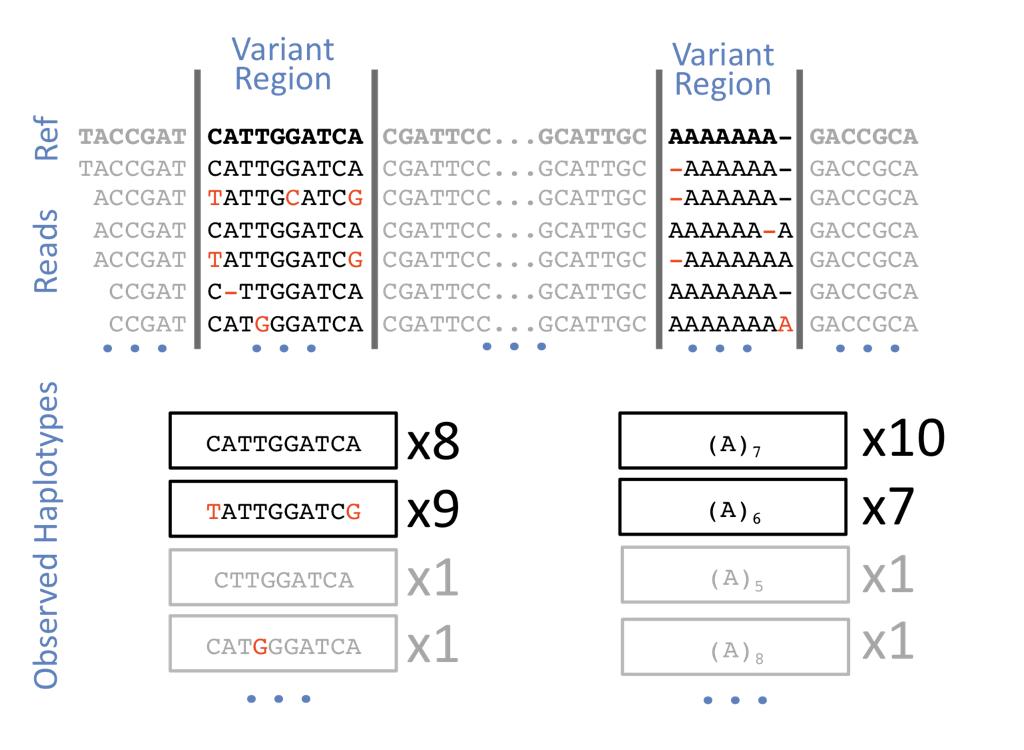
Now we need to start calling more SNPs on the indiviudals that have been mapped to the reference genome. I’m breaking this up into groups of individuals (200-300) across many populations. I’m also submitting this via sbatch rather than spinning up a srun /bin/bash approach… Failed.
Published
February 21, 2023
library( tidyverse )
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.4.4 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library( ggrepel )theme_set( theme_minimal() )
As a first pass through the data, I split individual sequences into different groups to run bwa. This works on an individual-by-individual basis and with 5% of each individuals genome takes roughly 30 minutes per individual (*1419 individuals …). Doing it in smaller groups allows me to run multiple instances at the same time. See the table here for the groups and current status. At the time of this writing, this process is still waiting on the final 6 groups of samples to be processed.
For a subset of the data, I did call SNPs at the same time that
Batching freebayes
To batch these I made a new directory called snpcalling and linked the bam files from a set of indiviudals in several different populations to this folder.
for bam in ../samples/O/*.bam;doln-s$bam;done
I then made a small runFreeBayes.sh run file that would